苯丙酮尿症(PKU)
(临床表现和治疗是重点掌握)
总述
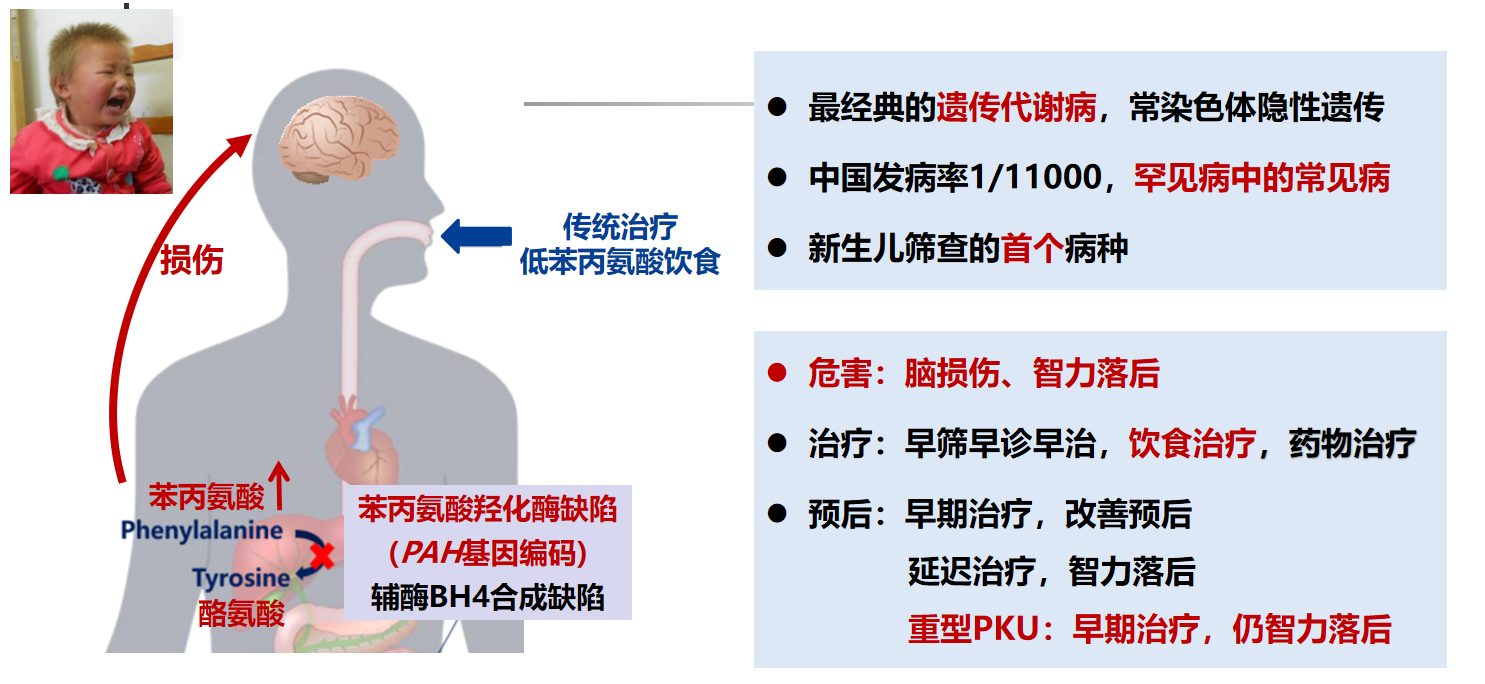
发病机制
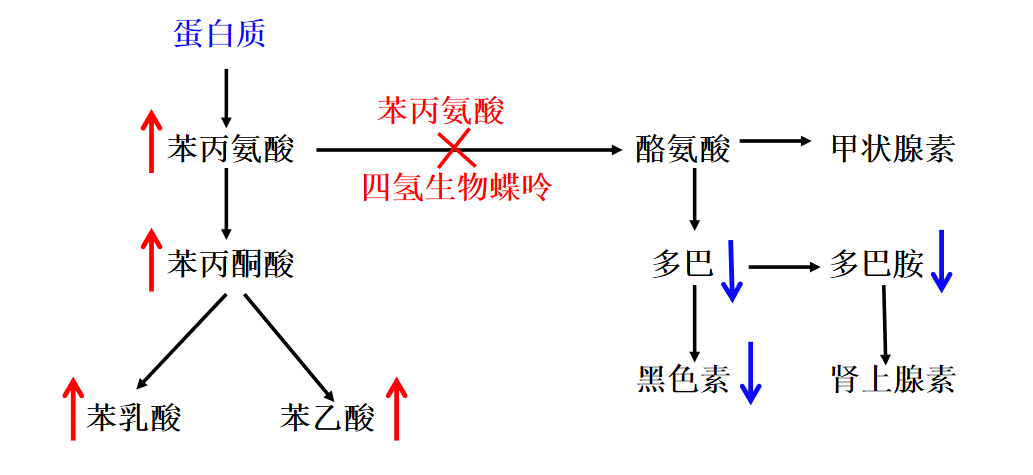
苯丙氨酸羟化酶、四氢生物蝶呤的缺失,导致苯丙氨酸和苯丙酮酸升高,多巴胺和黑色素降低
概念
➢ 高苯丙氨酸血症(hyperphenylalaninemia,HPA):血Phe≥120μmol/L且Phe/酪氨酸≥2
➢ 苯丙酮尿症(Phenylketonuria,PKU)是一种由于基因突变导致苯丙氨酸羟化酶活性降低或其辅酶四氢生物喋呤(BH4)缺乏,导致血苯丙氨酸(Phenylalanine,Phe)浓度增高的氨基酸代谢病,血Phe≥360μmol/L。
➢ 苯丙氨酸羟化酶缺乏症(PAH):
➢ BH4缺乏症
临床表现
经典型PKU
- 新生儿期无异常表现,2~3个月出现症状
- 呕吐、容易激惹、生长迟缓
- 头发由黑变黄、皮肤颜色浅淡
- 智能发育落后,小头畸形
- 尿、汗等分泌物有鼠臭味、皮肤湿疹
- 癫痫及婴儿痉挛、肌张力增高、腱反射亢进
- 多动、自残、攻击、忧郁
BH4缺乏症
- 具有PKU的临床表现
- 运动障碍,肌张力低下,眼睑下垂,眼震颤,吞咽困难,口水增多,嗜睡、
- 面无表情,反应迟钝,抑郁,失眠
- 顽固性抽搐,反复发热
- 智力严重落后
辅助检查
血Phe检测
HPA分型检测
BH4负荷试验
尿蝶呤谱检测
血红细胞二氢蝶啶还原酶活性检测
基因检测
HPA诊断
- 血Phe浓度≥120μmol/L(参考值20∼120)(>2 mg/dl)
- 血Phe/Tyr≥2.0(参考值0.2∼2.0)
- 除外其他原因导致的血Phe增高
鉴别诊断
- 希特林蛋白缺乏症:除Phe增高外,血瓜氨酸增高,伴或不伴蛋氨酸、Tyr增高,Phe/Tyr正常。
- 酪氨酸血症:血Tyr增高,Phe轻度升高/正常
- 肝功能损伤:多种氨基酸增高,Phe/Tyr正常
- 静滴氨基酸:多种氨基酸增高,Phe/Tyr正常
苯丙氨酸羟化酶缺乏症
尿蝶呤谱正常,二氢蝶啶还原酶活性正常,PAH基因突变
BH4缺乏症
6-丙酮酰四氢蝶呤合成酶(PTPS)缺乏症:血Phe增高,尿生物蝶呤降低,生物蝶呤%下降,PTS基因突变
二氢蝶啶还原酶(DHPR)缺乏症:血Phe增高,尿生物蝶呤及B%增高,DHPR活性降低,DHPR基因突变
治疗
PAH缺乏型PKU
➢ 对象:血Phe浓度<360μmol/L者不需要治疗;Phe浓度>360μmol/L者均应治疗
➢ 原则:控制天然蛋白质摄入,降低血Phe水平,维持血Phe浓度:
120~240μmol/L(≤1岁);
120~360μmol/L(1-12岁);
120~600μmol/L(>12岁)
➢ 饮食治疗:天然蛋白质:1~1.5g/Kg.d;无Phe营养粉:2~2.5g/Kg.d。
低Phe食品:蛋白粉、大米、面粉、饼干、调味料等
➢ 药物:BH4反应型者可单用BH4(5~20mg/kg.d) 或联合无Phe营养粉治疗
HPA治疗用营养粉及食品(营养粉、蛋白粉、低蛋白米)
➢ 血Phe浓度监测:<1岁每1~2周1次,1~12岁每2周~每月1次,>12岁每1~3个月1次
定期评价小儿生长发育及智能发育
饮食治疗至少至青春发育期,或终身治疗
BH4缺乏症
- 原则:降低血 Phe 浓度,补充神经递质
- 四氢生物蝶呤(BH4):PTPSD,1~10mg/Kg.d;DHPRD,10~30mg/kg.d
- 左旋多巴: 5~15mg/Kg.d,开始1~2mg/ Kg.d,以后每周增加1~2mg/Kg.d
- 5-羟色胺:5~10mg/Kg.d,开始1~2mg/ Kg.d,以后每周增加1~2mg/Kg.d
- 四氢叶酸:10~20mg/d(用于二氢蝶啶还原酶缺乏症)
预后
预后与血Phe浓度、治疗早晚、营养状况、治疗依从性等多种因素有关
新生儿筛查确诊、及时治疗的患者,大部分智力及体格发育正常
极少数患者即使早期筛查诊断、早期治疗,智能发育仍落后
极少数成年期存在认知、精神异常或社交能力落后等问题
产前诊断
- 父母再生孩子患病的概率为25%
- 父母再生孩子可以进行产前诊断,避免再生高苯丙氨酸血症患儿
(注:先证者基因突变位点必需明确)
➢ 孕17-20周羊水细胞基因检测
➢ 孕11-13周绒毛膜组织基因检测
➢ 无创基因基因检测(通过母亲血检测胎儿,今后发展的方向)
➢ 胚胎植入前单个细胞基因检测(试管婴儿,其他疾病的可能性)
小结
最常见的氨基酸代谢病,属于常染色体隐性遗传病,主要损伤大脑
新生儿筛查早诊断、早治疗、预后佳
有7种基因型,其中PAH及PTS基因较常见
是可治性遗传代谢病,特殊饮食或药物治疗,治疗及时,发育正常
可以产前诊断
思考题
HPA的病因?属于何种遗传方式?
HPA有哪些主要临床表现?
HPA的诊断标准?有几种类型?
HPA的主要治疗方法是什么?
HPA新生儿筛查方法?